AUNP-12 - A NOVEL PEPTIDE THERAPEUTIC TARGETING PD-1 IMMUNE CHECKPOINT PATHWAY FOR CANCER IMMUNOTHERAPY STRUCTURE ACTIVITY RELATIONSHIPS & PEPTIDE / PEPTIDOMIMETIC ANALOGS
Aurigene Discovery Technologies, the Bangalore-based biotech company, and Pierre Fabre, the second largest independent French pharmaceutical company, announced on February 11, 2014, a collaborative license, development and commercialization agreement giving Pierre Fabre worldwide rights (excluding India) to a new immune checkpoint modulator, AUNP-12, for the treatment of cancer. Aurigene will receive an upfront payment as well as milestone payments based upon continued development, regulatory progress, and product commercialization.[1]
AUNP-12, likely to be identical to the compound previously known under the codenames Aur-012, Aurigene-012, or Aurigene NP-12, is an inhibitor of the so-called PD-1 pathway, and will be in development for several cancer indications. It is so far the only peptide therapeutic in this pathway and could offer more effective and safer combination opportunities compared to current approaches,[2-4] e.g. antibodies such as Nivolumab (BMS), Lambrolizumab (Merck-3475), CT-011 (Curetech), MDX-1105 (BMS), MPDL3280 (GNE) and MEDI-4736 (Medimmune-AZ), or Amplimmune’s PD-L2-FC fusion protein.
PD-1, or Programmed cell death 1, is an immunoreceptor belonging to the CD28 family, and plays an important role in negatively regulating immune responses. The amino acid protein structure includes an extracellular amino acid IgV domain followed by a transmembrane region and an intracellular tail. PD-1 is expressed on the surface of activated T cells, B cells, and macrophages, and has two ligands, PD-L1 and PD-L2, which are members of the B7 family. PD-L1 is expressed on almost all murine tumor cell lines, whereas PD-L2 expression is more restricted and is expressed mainly by DCs and a few tumor lines.[5][6]
Blocking of PD-1 signaling pathways has been shown to result in restoration of defective immune cell functions in cancer and chronic infections. Recent advances in achieving highly durable clinical responses via inhibition of immune checkpoint proteins including PD-1 using antibodies or fusion proteins have revolutionized the outlook for cancer therapy. However, along with impressive clinical activity, severe immune-related adverse events (irAEs) due to the breaking of immune self- tolerance are becoming increasingly evident. Sustained target inhibition as a result of a long half-life (>15-20 days) and >70% target occupancy for months are likely contributing to severe irAEs observed in the clinic with antibodies targeting immune checkpoint proteins.[7]
In order to address the limitations of the current clinical candidates, Aurigene’s scientists focused on developing peptide-based immune checkpoint blockers with potent anti-tumor activity but with a shorter pharmacokinetic profile as a strategy to better manage severe irAEs.
Sequences of the extracellular domain of PD-1 critical for ligand-receptor interaction, overlapping or in close proximity to the known ligand binding regions,[6] were identified and served as starting points for the investigation of 7- to 30-mer peptides derived from human and murine PD-1 sequences, combined in a non-linear manner,[8] culminating in the discovery of AUNP-12 (a 29-mer).
Although the structure has not yet been released into the public domain, it is likely to correspond to one of the branched 29-mer peptides described in US Pat. Appl. 2011/0318373, and eventually to be identical to one the most active and best-described compounds, i.e. compound 8 (which is also taken as reference compound in Aurigene’s patent application, with an activity expressed as 100% (Figure 1, Table 1). Possible alternatives could be highly active N-acylated derivatives, such as 33, 34 or 83, although no mention is made in public disclosures of such a derivative.
|
FIGURE 1: Proposed structure of AUNP-12 (compound 8 of US 2011/0318373)
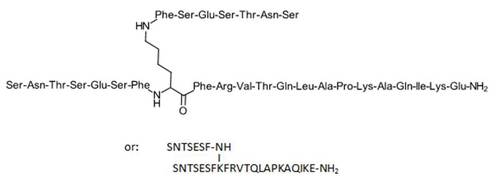
|
The series of compounds displays the following structure-activity relationships (Table 1):
- the C-terminal chain length of branched peptides is highly important for biological activity, as deletions (30, 31), or shorter analogs (41, 29) result in less active compounds
- modification of compound 8 by
- acylation on C-terminal lysine side chains by various long-chain aliphatic acids (34, 45, 33), or even acyl linkers including a potential Michael acceptor such as N-maleimide (83), susceptible to lead to irreversible binding by reaction with Cys thiol groups, is well tolerated and leads to some of the most active compounds of the series (33, 34, 83)
- acylation on the N-terminal Ser leads to loss of activity (16, 17, 20). Acylation of both N-terminal Ser restores some of the activity (21)
- unbranched peptides, including shorter chain derivatives (26, 27), and in particular the N-terminal heptapeptide 1 show surprisingly high activities
- other allowed modifications include
- derivatization of a C-terminal lysine with tripeptide side chains resulting from deletions in the main chain (35, 36)
- replacement of an arginine by an acylated lysine (40)
- replacement of the SNTSESF branch by a shorter, modified peptide sequence (37)
- peptides resulting from murine sequence fragments are usually less active (69, 91).
|
TABLE 1: Major Structure-Activity Relationships: activity (%) in mouse splenocyte proliferation assay(a)
| Compound n° (b) |
Sequence |
% |
| 1. Influence of C-terminal chain length of branched peptides(c): |
| 8 |
 |
100 |
| 30 |
 |
58,8 |
| 31 |
 |
11,3 |
| 41 |
 |
5,1 |
| 29 |
 |
31,1 |
| 2. Influence of side-chain and N-terminal acylations in branched peptides: |
| 34 |
 |
106 |
| 45 |
 |
68 |
| 83 |
 |
80,5 |
| 33 |
 |
98 |
| 46 |
 |
67 |
| 17 |
 |
32,9 |
| 16 |
 |
45,8 |
| 20 |
 |
27,8 |
| 21 |
 |
74,6 |
| 3. Influence of chain length in unbranched peptides: |
| 1 |
 |
96,5 |
| 27 |
 |
73,6 |
| 26 |
 |
86,6 |
| 43 |
 |
66 |
| 4. Examples of other modifications: |
| 36 |
 |
89,7 |
| 35 |
 |
81,2 |
| 40 |
 |
82 |
| 37 |
 |
89,6 |
| 69 |
 |
48 |
| 91 |
 |
69,4 |
(a): percent rescue with compounds screened at 100 nM concentration and compared with 100 nM compound 8 in mouse splenocyte proliferation CFSE based assay
(b): compound numbering as in patent application US 2011/0318373
(c): (-) denotes an amino acid deleted compared to the sequence of 8
(d): NMI = N-maleimide
(e): murine sequence |
|
In vitro and in vivo data confirm compound 8, and some of its acylated derivatives such as 34 and 83, as the most potent compounds of the series:
- compound 8 :
- displays an EC50 = 0.72 nM in the inhibition of binding PD1 to PD-L2 using hPDL2 expressing HEK293 cells, and an EC50 = 0.41 nM in a rat peripheral blood mononuclear cells (PBMC) proliferation assay using hPDL1 expressing MDA-MB231 cells. This corresponds well to the ‘sub-nanomolar potency in disruption of PD1-PDL1/2 interaction’ reported for AUNP-012[9]
- is equipotent to 83, but less potent than 34 and 45 in the restoration of human PBMC proliferation assay with recombinant hPDL1
- is equipotent to 34 in the mouse splenocyte proliferation rescue assay (mouse splenocyte proliferation inhibited by MD-MBA231 tumor cells expressing PDL1)
- is more potent than 34, which is equipotent to 83, in the IFNgamma production assay in a cytotoxic T lymphocyte assay
- inhibits by 44% tumor growth of B16F10 mouse melanoma cells injected subcutaneously in mice (5 mg/kg, subcutaneously once daily, 14 days)
- reduces lung metastasis of B16F10 cells injected iv. in mice (5 mg/kg, subcutaneously, once daily, 11 days), although being slightly less active than compounds 34 and 82,
- inhibits by 44% tumor growth of 4T1 cells injected orthotopically to mammary fat pad in mice (3 mg/kg, subcutaneously, once daily, 40 days). 10% of the animals treated with compound 8 showed complete regression and another 10% showed partial regression of tumor growth. Compound 8 treated animals showed a mean reduction in lung metastasis, measured after euthanasia, to the extent of >60%
- is active in vivo against E.coli sepsis
- in addition, compounds 34 and 83 are equipotent in reducing tumor burden in kidney injected orthotopically with Renal Carcinoma cells (5mg/kg, qd, 21 days).
Further in vivo studies revealed that AUNP-12/AUR-012 exhibits an excellent PK-PD correlation with sustained PD for >24 h. In preclinical models of melanoma, breast and kidney cancers, AUR-012/AUNP-12 showed superior efficacy compared to therapeutic agents currently used in the clinic in inhibition of both primary tumor growth and metastasis. Interestingly, dosing once in three days was equally efficacious as once a day dosing with no signs of overt toxicity and generation of neutralizing activity.[9]
Rescue of proliferation of immune cells analyzed upon stimulation with anti-CD3/anti-CD-28 indicated a complete rescue of CD4+ and CD8+ T cells. Interestingly, the proliferation of CD4+, Foxp3+ T cells was completely abolished with AUR-012/AUNP-12 treatment indicating a complete suppression of regulatory T cells. Sustained activation of circulatory immune cells and their ability to secrete IFN-γ up to 72 h indicate that pharmacodynamic effects persist even after the clearance of the compound in animal models, thus supporting a dosing interval of up to 3 days. In models of melanoma, breast, kidney and colon cancers, AUR-012/AUNP-12 showed efficacy in inhibition of both primary tumor growth and metastasis. Additionally, anti-tumor activity of the compound in a pre-established CT26 model correlated well with pharmacodynamic effects as indicated by intratumoral recruitment of CD4+ and CD8+ T cells, and a reduction in PD1+ T cells (both CD4+ & CD8+) in tumor and blood. In 14-day repeated dose toxicity studies, AUR-012/AUNP-12 was well tolerated at 100 times the efficacious doses.[7]
|
Further developments
Interestingly, compound 1, a short heptapeptide, displays a high percentage of inhibition in the mouse splenocyte assay. Its sequence corresponds to the so-called BC loop,[6] i.e. amino acids 24-30 of the extracellular domain of PD1.
This has led Aurigene scientists to develop several series of modified short chain peptides and peptidomimetics of this sequence with significant levels of activity:
1. 7- to 8-mer peptides including a central linker[10]
The most active compound in a series of linear hepta- and octapeptides derived from the BC loop and including a central linker of various nature and length between residues corresponding to amino acids 28 and 29 is compound 1 (Table 2).
|
TABLE 2: Percentage of activity in mouse splenocyte proliferation assay
| Compound n° (a) |
Structure |
% rescue (b) |
| 1 |
 |
87 |
(a): Compound numbering as in patent application WO 2012/168944
(b): compared to rescue by splenocytes stimulated by anti-CD3 and anti-CD28 antibody (calculated from the corresponding fold induction values) |
|
The structure activity relationship of the series tolerates substituents on the Phe aromatic ring, N-terminal acetylation, and D-amino acids in several positions, including an all-D analog of 1. Compound 1 exhibits a 4-fold induction (no concentration given), compared to a 4.6-fold induction with stimulated splenocytes, in the mouse splenocyte proliferation assay inhibited by MD-MBA231 tumor cells expressing PDL1, and is active in vivo in a lung metastasis model of B16F10 melanoma in mice, showing a 64% reduction in metastasis at 5 mg/kg (subcutaneous, once daily, 14 days).
2. 7- to 9-mer cyclic peptides[11]
Hepta- to nonapeptides derived from the same PD1 BC loop as above, and rigidified by connecting side chains of lysine and glutamate amino acids or by cyclizing the N-terminal Ser and a C-terminal Arg through amide bonds to give lactams, lead to compounds which inhibit PD1/P-L1 interaction as measured by the mouse splenocyte proliferation assay (Table 3). The structure activity relationships also tolerate the replacement of the N-terminal Serine and other amino acids in central positions of the peptides by various amino acids (Table 3).
|
TABLE 3: Activity in mouse splenocyte proliferation assay
|
The most active compounds exhibit a more than 4-fold induction (no concentration given), compared to a 5,5-fold induction with stimulated splenocytes. Compound 2 is active in vivo in a lung metastasis model of B16F10 melanoma in mice, showing a 54% reduction in metastasis at 5 mg/kg (subcutaneous, once daily, 14 days), compared to a 48% reduction for Taxol at the same dose.
3. Tripeptide peptidomimetics[12]
Tripeptide peptidomimetics including diacylhydrazine and urea linker moieties potently inhibit both PD-L1 and PD-L2 binding to PD1 as measured in the splenocyte effector function assay by monitoring IFNgamma release. For both ligands, EC50 values are in the nanomolar range. Structure activity relationships appear to favor a C-terminal acid over the amide function.
Compound 2 exhibits in vivo efficacy by reducing tumor growth by 46% in a CT-26 colon cancer model in mice at a dose of 3 mg/kg (25 days). It is also active against Pseudomonas aeruginosa in a lung infection model in mice at 10mg/kg (three times a day, 11 days).
|
TABLE 4: EC50 values in mouse splenocyte effector function by monitoring IFNgamma release
|
Other small molecule approaches
A group at Harvard has published on the discovery of small molecule modulators of PD-1, derived from the known compounds sulfamonomethoxine and sulfamethizole, which are both active as antagonists in an interferon gamma release assay in transgenic mouse T cells that constitutively express PD-1 (Fig 2).[13] The activity of these compounds is however relatively weak, in the micromolar range. No further biological data have been reported so far.
|
FIGURE 2: Small molecule antagonists of the PD-1 pathway
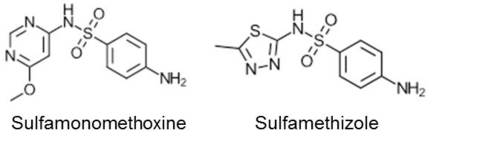
It is however likely to see further developments in the search for small molecule antagonists of the PD-1 pathway.
|
Conclusion and outlook
Blocking the PD-1 pathway has become a highly active area in cancer immunotherapy,[14] with antibodies to PD-1 and its ligands PD-L1/L2 in advanced clinical studies, and combination studies with other anticancer agents ongoing.
The more recent addition of peptidic antagonists of PD-1 to PD-L1/L2 binding such as AUNP-12 opens the possibility to reduce severe immune-related adverse events due to prolonged target occupancy by modulating the half-life of the drug. The chemical structure of AUNP-12 remains to be confirmed, but could correspond to compound 8 as described above.
It remains to be seen how a 29-mer peptide will compare to small peptides and peptidomimetics, which are significantly easier and cheaper to produce, as well as to other small molecule approaches which are, however, currently lacking the high activity and probably also selectivity of larger molecules.
|
Bibliography
[1] Aurigene & Pierre Fabre, "Aurigene Discovery Technologies Limited And Pierre Fabre Announce A Licensing Agreement For A New Cancer Therapeutic In Immuno-Oncology: AUNP12, An Immune Checkpoint Modulator Targeting The PD-1 Pathway," 11 Feb 2014: http://aurigene.com/news/aurigene-discovery-technologies-limited-and-pierre-fabreannounce-a-licensing-agreement-for-a-new-cancer-therapeutic-in-immuno-oncology-aunp12-an-immune-checkpoint-modulator-targeting-the-pd-1-pathwa/. [Accessed 21 Feb 2014].
[2] D. B. Flies, B. J. Sandler, M. Sznol and L. Chen, "Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy," Yale Journal of Biology and Medicine, vol. 84, pp. 409-421, 2011.
[3] M. Sznol, "Cancer immunotherapies: novel treatment opportunities and their implications (Cancer Crosslinks, Oslo, Norway)", 23 January 2014: http://www.oslocancercluster.no/LinkClick.aspx?fileticket=i_7m5OTLv0U%3D&tabid="133". [Accessed 24 February 2014].
[4] T. P. McGarty, "PD-1: Another immune inhibition for T cell therapeutics for melanoma," June 2013: http://www.telmarc.com/Documents/White%20Papers/96%20PD-1.pdf. [Accessed 24 February 2014].
[5] "Programmed Cell Death 1": http://en.wikipedia.org/wiki/Programmed_cell_death_1, [Accessed 21 Feb 2014].
[6] E. Lázár-Molnár, Q. Yan, E. Cao, U. Ramagopal, S. G. Nathenson and S. C. Almo, "Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2," Proc. Natl. Acad. Sci. USA, vol. 105, no. 30, pp. 10483-10488, 2008.
[7] P. Sasikumar, R. Shrimali, S. Adurthi, R. Ramachandra, L. Satyam, A. Dhudashiya, D. Samiulla, K. B. Sunilkumar and M. Ramachandra, "A novel peptide therapeutic targeting PD1 immune checkpoint with equipotent antagonism of both ligands and a potential for better management of immune-related adverse events," Journal for ImmunoTherapy of Cancer, vol. 1, no. Suppl 1, O24, 2013.
[8] P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Vemula, L. K. Satyam, K. Subbarao, K. R. Shrimali and S. Kandepudu (Aurigene Discovery Technologies Ltd, Bangalore, India), "Immunosuppression modulating compounds", US Patent application US 2011/0318373, 29 Dec 2011.
[9] P. G. Sasikumar, L. K. Satyam, R. K. Shrimali, K. Subbarao, R. Ramachandra, S. Vadlamani, A. Reddy, A. Kumar, A. Srinivas, S. Reddy, S. Gopinath, D. S. Samiulla and M. Ramachandra, "Demonstration of anti-tumor efficacy in multiple preclinical cancer models using a novel peptide inhibitor (Aurigene-012) of the PD1 signaling pathway," Cancer Research, vol. 72, no. 8 Suppl. 1, Abstract 2850, 2012.
[10] P. G. N. Sasikumar, M. Ramachandra, S. K. Vadlamani, K. R. Shrimali and K. Subbarao, "Therapeutic compounds for immunomodulation" (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO 2012/168944, 13 Dec 2012.
[11] P. G. N. Sasikumar and M. Ramachandra, "Immunomodulating cyclic compounds from the BC loop of human PD1" (Aurigene Discovery Technologies Ltd, Bangalore, India), PCT Patent Application WO/2013/144704, 3 Oct 2013.
[12] P. G. N. Sasikumar, M. Ramachandra and S. S. S. Naremaddepalli, "Peptidomimetic compounds as immunomodulators" (Aurigene Discovery Technologies Ltd, Bangalore, India), US Patent Application US 2013/0237580, 12 Sep 2013.
[13] A. H. Sharpe, M. J. Butte and S. Oyama (Harvard College), "Modulators of immunoinhibitory receptor PD-1, and methods of use thereof", PCT Patent Application WO/2011/082400, 7 Jul 2011.
[14] M. Cordingley, "Battle of PD-1 blockade is on", February 7, 2014 : http://discoveryview.ca/battle-of-pd-1-blockade-is-on/ [Accessed 25 February 2014].
|
|
|








